How reliable are protein structures determined by electron microscopy?
03-03-2017
Writer(s): Daisuke Kihara
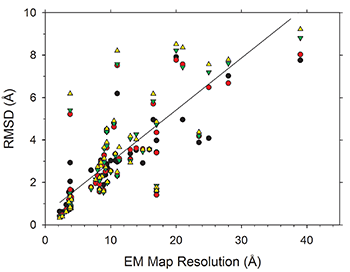
An increasing number of biomolecular structures are solved by electron microscopy (EM). However, the quality of structure models determined from EM maps vary substantially. To understand to what extent structure models are supported by information embedded in EM maps, Lyman Monroe and Dr. Genki Terashi, together with Prof. Kihara, used two computational structure refinement methods to examine how much structures can be refined using a dataset of 49 maps with accompanying structure models. The extent of structure modification as well as the disagreement between refinement models produced by the two computational methods scaled inversely with the global and the local map resolutions. Specifically, root mean square deviation (RMSD) of modification to a structure was roughly one-third of its global map resolution and roughly half of its local map resolution. The results indicate that the observed discrepancy between the deposited map and the refined models is due to the lack of structural information present in EM maps and thus these annotations must be used with caution for further applications.
The paper was released online on Structure.
Paper: http://www.cell.com/structure/fulltext/S0969-2126(17)30035-7
Variability of Protein Structure Models from Electron Microscopy. Lyman Monroe#, Genki Terashi#, Daisuke Kihara (# co-first author), Structure, Published online on March 2, 2017. DOI: http://dx.doi.org/10.1016/j.str.2017.02.004
Contact: Daisuke Kihara http://kiharalab.org