Mechanistic insights into Lowe syndrome may lead to new therapies
12-10-2013

Despite being described in the early 1950s, the underlying mechanism of Lowe syndrome (LS) is still poorly understood. This devastating developmental disease is characterized by bilateral cataracts at birth, mental retardation and kidney malfunction. Unfortunately, LS children die early, mostly due to renal failure. Although the affected gene, OCRL1 (that encodes the Ocrl1 enzyme), was identified more than 20 years ago, the process which causes the dysfunctions characteristic of LS is not yet clear. A team led by Dr. R. Claudio Aguilar recently discovered that cells of LS patients have defects in the assembly of the primary cilium, a hair-like sensory organelle present in many cells. As a result, sensory signaling pathways are defective, which would lead to abnormalities in multiple organs. Dr. Aguilar's group also demonstrated LS-like cilium abnormalities in ocrl1-deficient zebrafish. Importantly, function could be restored by reintroducing normal enzyme.
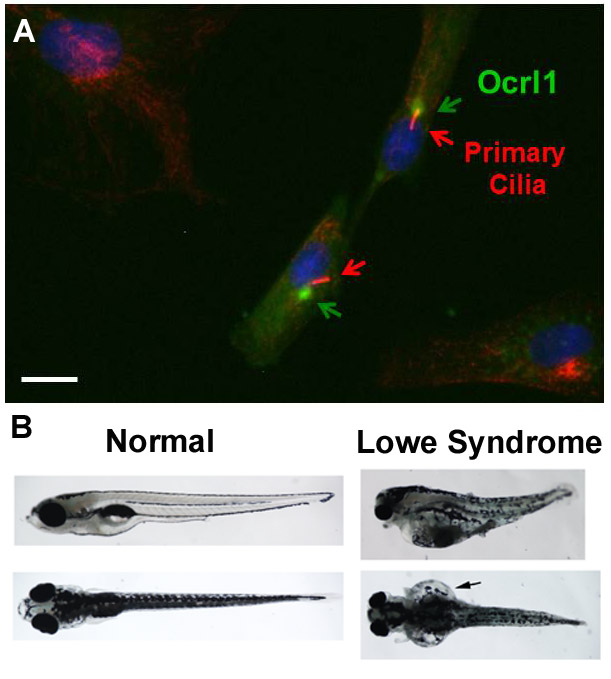
In the study published last year in Human Molecular Genetics, the authors demonstrated that the Ocrl1 enzyme is involved with the process of protein transport, which in turn is required for the assembly of theprimary cilium. This finding establishes the mechanism by which Ocrl1-deficiencies lead to primary cilia abnormalities and therefore, to LS patient multi-organ symptoms. Importantly, this work also shows that LS is not an isolated disease, rather it is associated with a broader group of disorders collectively known as ciliopathies. This novel view of the disease could have a major impact in the field and ultimately on patients' well-being. Breakthroughs in terms of mechanistic insights or novel therapeutic approaches arising in the field of ciliopathies could be immediately capitalized upon by LS researchers and lead to viable treatment options for LS.
This research was done primarily at Purdue University by Biology graduate students Brian Coon and Kayal Madhivanan, with contributions by others from Purdue, University College London and the University of Manchester, and it was supported by a grant from the Lowe Syndrome Trust.
Coon BG, Hernandez V, Madhivanan K, Mukherjee D, Hanna CB, Barinaga-Rementeria Ramirez I.,Lowe M, Beales PL, Aguilar RC. The Lowe syndrome protein OCRL1 is involved in primary cilia assembly.Hum Mol Genet 21: 1835–1847, 2012.
https://hmg.oxfordjournals.org/content/21/8/1835
Writer: Karen Wiggins, 49-44749, wiggins@purdue.edu.